Flex-EM 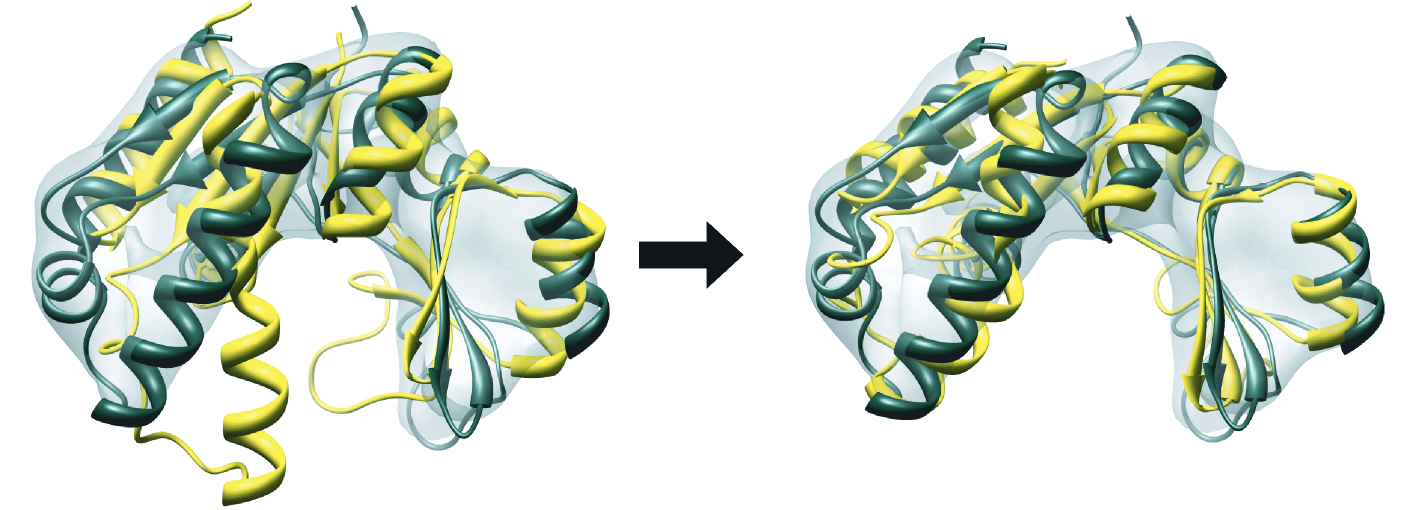
Fitting and
refinement of atomic structures guided by cryoEM density.
Please cite: M. Topf, K. Lasker,
B. Webb, H.
Wolfson,
W. Chiu, A. Sali. Structure 16, 295-307, 2008

Other citations: A.P. Joseph, S. Malhotra, T. Burnley, C. Wood, D.K. Clare, M. Winn, M. Topf. Methods 100, 42-49, 2016
The method includes a rigid
fitting
stage
followed by a refinement stage. Rigid
fitting can
be performed with Mod-EM
or any other rigid fitting method. The refinement stage starts with
the components rigidly fitted in the
approximate positions in the map. Two methods are available:
conjugate gradients minimization (CG) and simulated annealing molecular
dynamics
(MD). To use it:
A. Download the following
scripts:
B. Edit the following files:
flex-em.py (below INPUT PARAMETERS):
1. Define the mode of
optimization
(optimization='MD' or optimization='CG').
2. Set the input parameters of the atomic structure (PDB format) that
you want to fit and the EM map (MRC or XPLOR format).
3.
Specify the path of your work directory.
4. For CG optimization: specify the number of CG runs (eg, num_of_runs=2)
and the number of the initial directory in which you want the
runs to be found (eg, init_dir=1
will produce the results in the directories
1_cg, 2_cg).
5. For MD optimization: specify the number of MD
iterations (eg, num_of_iter=4)
and the number of the directory in which you want
the runs to be found (eg, init_dir=1
will produce the results in the directory
1_md).
Important Notes:
- If your origin is in pixels (e.g. from Chimera) please multiply by -1*voxel size.
- If you have chain breaks in your structure keep the
TER line in your PDB input file (otherwise the program will connect
your chains).
- If you know the approximate
initial positions of your components running the MD
refinement is recommended.
- If you are not sure
about the approximate positions of the components, it is recommended:
(a) First run the CG minimization multiple times
(eg, num_of_runs
= 20);
(b) Cluster
the different "fits";
(c) Once you have selected your ensemble of representative
"rigid
fits" you can start an MD refinement for each of them (using
each as an initial file for the MD optimization
(eg, input_pdb_file='final1_cg.pdb'
).
- For high-resolution refinement (better than ~4.5 Å) change the cap_shift in flex-em.py
. SSE restrained: cap_shift=0.15, all-atom refinement: cap_shift=0.10.
rigid.txt:
Define the rigid bodies using residue numbers. The format of
this file is the following:
- Comment lines begin with '#'
(eg, describing what kind of a rigid body : '#domain', '#helix', '#beta').
- Other lines: each line describes one rigid body by specifying the
initial and final residue of each of the segments in that rigid body
(eg, '2 6 28 30' means
that residues 2-6 and 28-30 will be included in the same rigid body).
- For multi-chain models: each line describes one rigid body by specifying the
initial and final residue of each of the segments, along with their chain IDs
(eg, '2:A 6:A 28:B 30:B' means
that residues 2-6 from chain A and 28-30 from chain B will be included in the same rigid body).
- For including ligands and other hetero-atoms (HETATM), the molecule number
and chain ID can be added to the rigid body line in the same format (but it should not form a separate rigid body).
(eg, '100:A 100:A' means
that the HETATM 100 from chain A will be considered as a rigid body).
- Only in CG mode: if
you add 'nr'
after the last segment (eg, '2 6 28 30
nr') the rigid
body will not be randomized prior to the running of
the optimization.
Important Notes:
- For a better description of your rigid bodies please use RIBFIND (see: Pandurangan & Topf, J Struct Biol 2011 ). The program will automatically produce your rigid.txt file.
C. Run the program (using MODELLER-9v2
or later):
mod9v9 flex-em.py > flex-em.log
The
refined structures will be found in the specified directory as
described above.
Flex-EM can be applied to
both experimental atomic structures and
homology models. tutorial:
flex_em_tutorial.pdf.
--------------------------------------------------------------------------------
Flex-EM benchamrk is available for download here.
- To minimize bias, the benchmark density maps were not
produced with Mod-EM,
but with pdb2vol in SITUS (Wriggers, et al. 1999),
which uses a
different Gaussian smoothing
technique.
- Cα RMSDs were calculated for the benchmark using the
superpose method in MODELLER-9v2.
For questions, please contact Maya Topf at
maya.topf@cssb-hamburg.de